
Current research activities and major results
3D Domain swapping and amyloidogenesis
Amyloid-forming proteins attract attention because of their role in the pathogenesis of a number of diseases, like Alzheimer's disease and the prionoses. The pathophysiological processes involve abnormal conformational changes, followed by aggregation. Human cystatin C (HCC), a potent and abundant inhibitor of cysteine proteases, changes its structure on prolonged incubation to form inactive dimers. The tendency to oligomerize may explain why HCC forms amyloid deposits in the brain arteries at advanced age. Formation of HCC amyloid is accompanied by the presence of HCC dimers in the cerebrospinal fluid. The amyloidogenic property of HCC is drastically amplified in the endemically occurring L68Q mutant that causes amyloidosis and brain hemorrhage leading to death in young adults. We have reported, for the first time, the crystal structure of HCC and demonstrated that the protein aggregates to form dimers through an exchange of structural units. This phenomenon of 3D domain swapping is a mechanism for oligomerization of monomeric proteins. The same mechanism of 3D domain swapping has been found for N-truncated HCC, which is the dominating material isolated from cystatin C deposits of patients suffering from HCC amyloidosis. The structure of 3D domain-swapped HCC dimers suggests possible mechanisms of amyloid fibril formation and explains the role of the L68Q mutation. It also has implications for other diseases involving conformational pathologies. In recent experiments, we have demonstrated that by strategic placement of cysteine residues in the HCC sequence, we can introduce new disulfide bridges, allowing red-ox control of domain swapping, and in consequence - control of dimerization, oligomerization, and amyloid fibril formation.
Cysteine proteases and their inhibitors

Several serious diseases related to tissue degeneration, such as osteoporosis or muscular dystrophy, are linked to abnormalities in the functioning of cysteine proteases. There is a variety of natural inhibitors of these enzymes, ranging from sizeable proteins, such as cystatins, to relatively small molecules, such as E-64. The latter compound contains an oxirane ring that blocks the enzyme by forming a covalent bond with the catalytic thiol group. We have studied a number of complexes of the model cysteine protease papain with synthetic inhibitors containing a peptide sequence modeled after the binding epitope of HCC, and a reactive group, such as oxirane or diazomethylketone. Cysteine proteases are also involved in pathogen invasion. We have determined the crystal structure of the wild-type zymogen (known as superantigen B) of the enzyme from the highly virulent bacterium Streptococcus pyogenes. In another project, we focus on the cysteine protease inhibitor, called chagasin, from the protozoan responsible for Chagas disease, Trypanosoma cruzi. Although chagasin has the same size as cystatins, our crystal structure has demonstrated that it has a completely unrelated fold. Nevertheless, the enzyme-binding epitope has the same three-loop architecture, as revealed by the crystal structures of inhibitory complexes between chagasin and the human enzymes cathepsin L and B and the model cysteine protease papain.
Retroviral enzymes
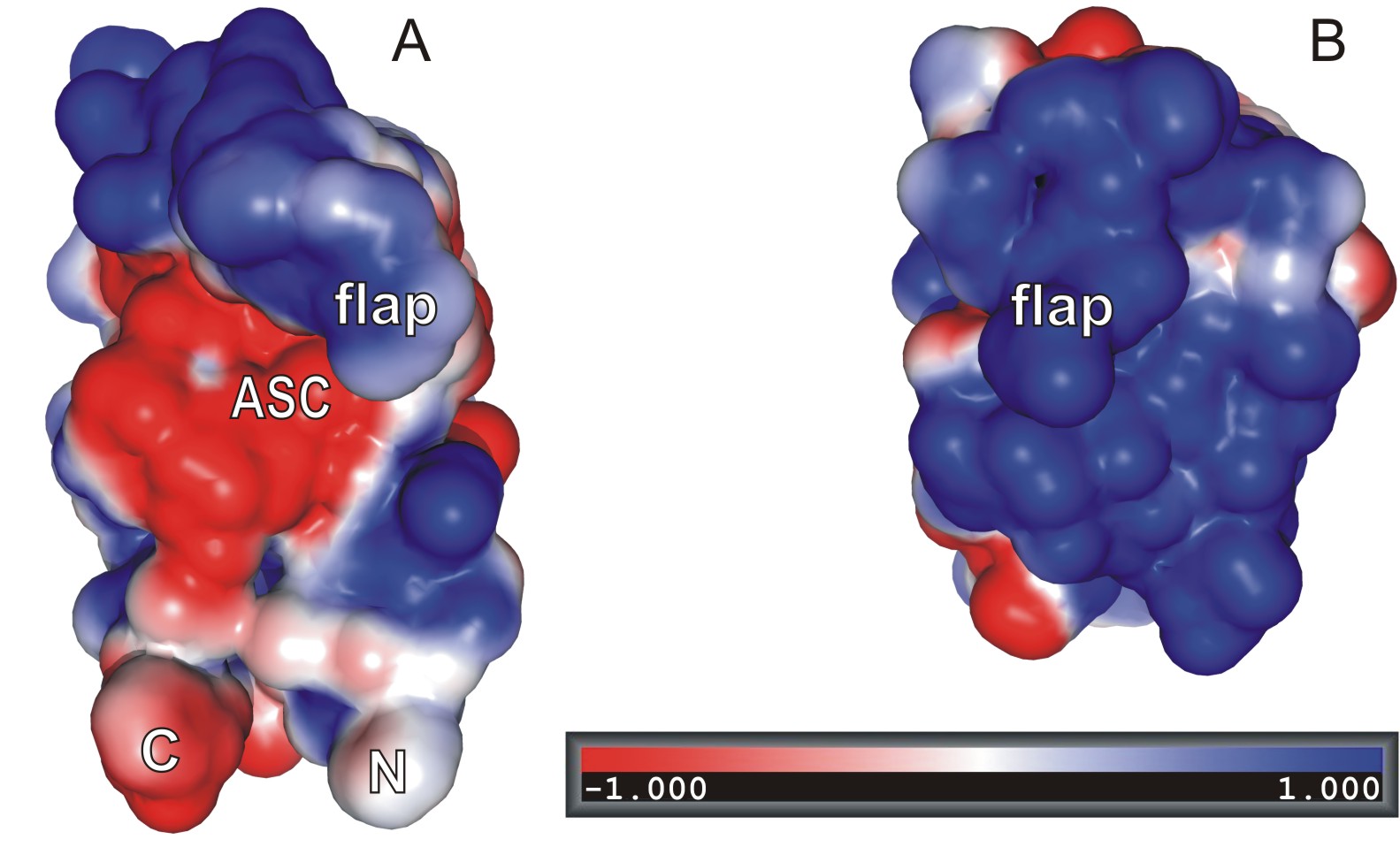
Retroviral protease is a key enzyme in the replication cycle of the HIV-1 retrovirus, the causative agent of AIDS. Since the determination of the three-dimensional structure of the enzyme (in a collaborative project with Dr. Alex Wlodawer, NCI, USA) and the discovery that it is a symmetric, homodimeric aspartic protease, it has become the main target in the rational design of drugs for the treatment of AIDS. Our current interest in this field focuses on new-generation active-site inhibitors of HIV-1 protease as potential drugs, on complexes of the enzyme with autodigestion products, and on proteases from other retroviruses. The recently determined structure of the enzyme from the leukemia-causing HTLV virus has revealed important differences in the active-site architecture between the HIV-1 and HTLV enzymes, thus explaining the failure of successful anti-AIDS drugs in the treatment of HTLV infections. Another retrovirus of interest is Mason-Pfizer Monkey Virus (M-PMV), which causes AIDS-like syndrome in rhesus monkeys. M-PMV protease can be purified and crystallized as a monomer, and its structure (B; compare it with that of HIV-1 protease protomer extracted from the dimeric context, A) is an important target for the design of dimerization inhibitors that could be developed as a new-generation antiretroviral drugs. The structure of M-PMV protease has been solved in an unprecedented manner, using -- as a molecular-replacement probe -- a model generated in a "citizen-science" experiment by players of the Foldit online game. We have also been studying the structure of retroviral integrase, which is responsible for the integration of the retroviral genetic material into the host cell genome. In particular, we have determined, again in a collaborative project with Dr. Alex Wlodawer, the structure of the catalytic domain in active conformation and in complex with divalent metal cations.
Antileukemic bacterial asparaginases

The interest in periplasmic bacterial L-asparaginases has been instigated by their antileukemic activity. Asparaginases of this type, for example Escherichia coli L-asparaginase II (EcAII), are homotetramers with four active sites. Each active site is created by amino acids from two monomers, including amino acids form conserved motifs. The mechanism of the asparaginase reaction is not fully understood. It could be a variant of the reaction catalyzed by serine proteases, but with a threonine in the role of the nucleophilic serine. The other residues of the putative catalytic triad could be D90 and K162. To view an electronic poster on the dynamics aspects of the L-asparaginase catalytic center click here . The antitumor activity of these enzymes is the effect of their high affinity for the substrate. Depletion of L-asparagine in the circulating pools starves the tumor cells, which have reduced levels of L-asparagine synthesis. We have studied a number of EcAII mutants to shed more light on the enzymatic mechanism of these enzymes. Recently, we have discovered that EcAII binds zinc cations, which may be of importance in its role as a drug.
Plant-type asparaginases

In plants, L-asparagine is the most abundant metabolite for the storage and transport of nitrogen that is utilized in protein biosynthesis. Asparagine hydrolysis in plants is catalyzed by asparaginases with no homology to the bacterial-type enzymes. The most studied enzymes in this class, from legume plants, are involved in metabolic pathways connected with assimilation of atmospheric nitrogen. We have cloned, sequenced, expressed, and crystallized the enzyme from Lupinus luteus (LlA). We have also shown that the E. coli genome encodes a highly homologous enzyme, EcAIII. The structure of EcAIII has been solved demonstrating that it is an N-terminal nucleophile (Ntn) hydrolase that undergoes autoproteolytic activation to liberate the N-terminal threonine (subunit beta) nucleophile. The active protein is an (alpha-beta)2 heterotetramer. The structure and maturation pattern place EcAIII in one class with aspartylglucosaminidases. However, enzymatic and kinetic studies show that both LlA and EcAIII are predominantly active as isoaspartyl aminopeptidases, and that their L-asparaginase activity is of secondary importance. In this light, LlA and EcAIII gain in relevance as enzymes responsible for the degradation of malformed proteins, in which a peptide bond occurs at the side chain of asparagine or aspartic acid.
Proteins from plant-bacterium symbionts and pathogens


Symbiosis between legume plants and nitrogen-fixing bacteria depends on exchange of precise molecular signals. This process results in the formation of root nodules in which the bacteria assimilate atmospheric nitrogen. In addition to plant signals, bacterial signals, called Nod Factors (NF), are also necessary. The unique NF biosynthetic pathway includes about a dozen enzymes whose structure is very poorly understood. We have cloned, overexpressed, and crystallized (also in selenomethionyl form) two rhizobial enzymes form this pathway (NodZ and NodS) and determined their structure by the MAD method. NodZ catalyzes fucosylation of the NF chitooligosaccharide (COS) molecule. The crystal structure reveals a two-domain folding pattern, with one of the domains folded according to Rossmann motif. This is consistent with the fact that GDP-fucose is the sugar donor in the fucosylation reaction. NodS N-methylates the non-reducing unit of the COS substrate, using SAM (S-adenosyl-L-methionine) as methyl donor. Our structures show that the N-terminal part of the enzyme gets ordered on SAM binding, creating at the same time a deep canyon for COS docking. The two substrates meet at an orifice leading to the SAM-cavity, where the NH2 nucleophile of COS attacks the CH3 group of SAM in the methyl-transfer reaction. Regulation of SAM-dependent methylations occurs via the action of SAHase, which decomposes SAH (S-adenoslyl-L-homocysteine), a byproduct of the methylation reaction. Originally we solved the crystal structures of SAHases from legume/Bradyrhizobium symbionts in complex with different ligands related to the SAHase reaction. Next we focused our attention on enzymes from extremofiles and pathogenic organisms.
Plant pathogenesis-related and hormone-binding proteins


The pathogenesis-related class 10 (PR-10) proteins have been detected in nearly all studied
plants but never in non-plant organisms. The PR-10 class includes numerous homologs with expression patterns responding to
pathogens and other stress factors. Tree pollen allergens are also in this class, similarly to the distantly related
cytokinin-specific binding proteins (CSBP). In contrast to CSBP, the function of classic PR-10 proteins remains unknown
despite their high cytosolic content. We have determined the crystal structure of a CSBP protein in complex with the plant
hormone zeatin, confirming that its fold is consistent with PR-10 classification. This fold consists of a seven-stranded
antiparallel beta-sheet wrapped around a long C-terminal helix. Between the sheet and the helix there is a large internal
cavity, where the ligands are bound. The C-terminal helix is unusual since it shows high sequence variability and geometrical
distortions. These distortions determine the shape and size of the cavity and may, therefore, control the ligand specificity of
the protein. Recently, we also managed to crystallize a classic PR-10 protein in complex with zeatin. In both complexes there
are multiple copies of the hormone in the binding cavity (CSBP - two, PR-10 - three) but the modes of binding are
different.

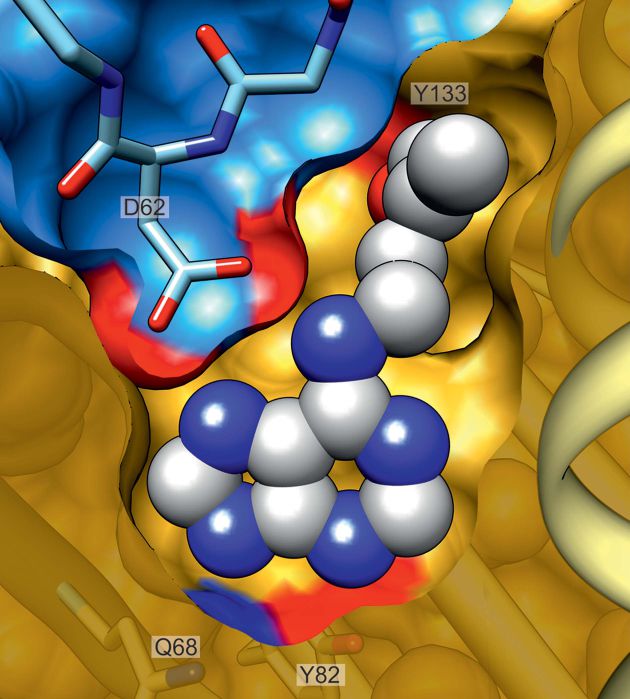 On the other hand, the St John's wort protein Hyp-1, purportedly responsible for the biosynthesis of the
pharmacologically active hypericin, seems to be just a PR-10 protein, but it has an unusal pattern of three binding sites, illustrated here by a complex with the fluorescent dye ANS
(8-anilino-1-naphthalene sulfonate). In another study, we have shown that Nodulin 13 from Medicago truncatula (MtN13, involved in root nodulation during symbiosis with
nitrogen-fixing bacteria) is a PR-10 protein binding, upon its dimerization, different cytokinins in a very specific way, with 2:2 stoichiometry. Recently, we discovered that
plant PR-10 are very good binders of melatonin, which is a newly emerging phytohormone. Moreover, we have structural evidence confirming that PR-10 proteins play a mediating
role in crosstalk of melatonin with other plant hormones and metabolites.
On the other hand, the St John's wort protein Hyp-1, purportedly responsible for the biosynthesis of the
pharmacologically active hypericin, seems to be just a PR-10 protein, but it has an unusal pattern of three binding sites, illustrated here by a complex with the fluorescent dye ANS
(8-anilino-1-naphthalene sulfonate). In another study, we have shown that Nodulin 13 from Medicago truncatula (MtN13, involved in root nodulation during symbiosis with
nitrogen-fixing bacteria) is a PR-10 protein binding, upon its dimerization, different cytokinins in a very specific way, with 2:2 stoichiometry. Recently, we discovered that
plant PR-10 are very good binders of melatonin, which is a newly emerging phytohormone. Moreover, we have structural evidence confirming that PR-10 proteins play a mediating
role in crosstalk of melatonin with other plant hormones and metabolites.
Insect hormone-binding proteins

The complicated insect development program is orchestrated by hormones, such as Juvenile Hormone (JH). This very labile and highly hydrophobic molecule is transported in the insect hemolymph by a specialized protein called Juvenile Hormone Binding Protein (JHBP), as a 1:1 complex. Recently, we solved the structure of this mysterious protein from Galleria mellonella revealing its hot-dog-like fold, in which a long alpha-helix is almost completely wrapped in a highly curved beta-sheet. The same folding motif can be found in two low-homology human proteins, which also bind hydrophobic ligands, but it is present there in tandem repeats, indicate gene duplication. Surprisingly, the relatively small JHBP protein contains two hydrophobic pockets, one at each pole. Analysis of the interior of those pockets together with other biochemical experiments leaves no doubt as to which of them harbors the hormone binding site. So far, it was not possible to obtain crystalline JHBP-JH complex. Every time we try to soak the hydrophobic JH molecule into the JHBP crystals - they crack, hinting that there is a significant conformational rearrangement of the protein molecule on hormone binding.
Nuclear receptors

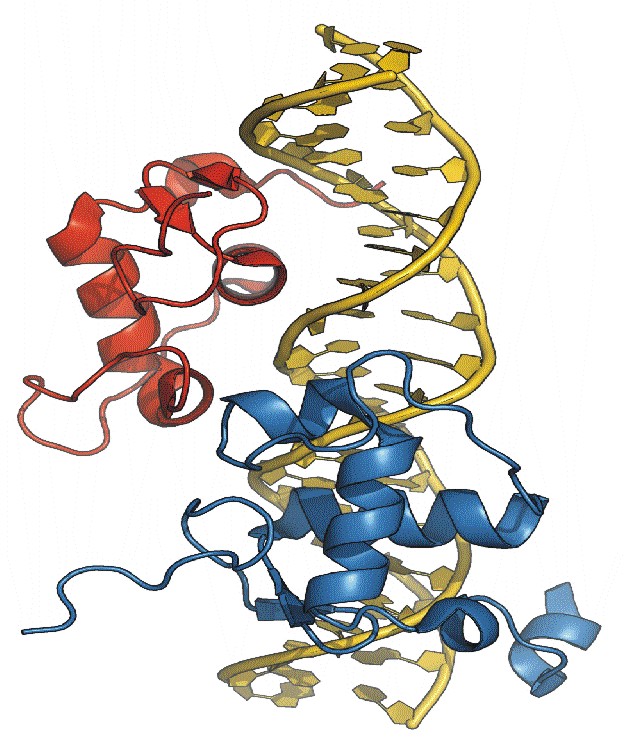
The functional nuclear receptor for the steroid hormones responsible for molting and metamorphosis in insects is an unusual heterodimeric molecule. The crystal structure of the DNA-binding Domains (DBDs) of the two partners (EcR and Usp) in complex with their natural DNA response element, hsp27, reveals a novel element (an alpha-helix) in the C-Terminal Extension (CTE) of the EcRDBD domain. The location of this helix in the minor groove of the DNA target does not match any of the locations reported previously for nuclear receptors. Mutational analyses suggest that this alpha-helix is a component of an EcR-box, a novel element indispensable for DNA-binding.
Structural chemistry of exotic nucleic acids

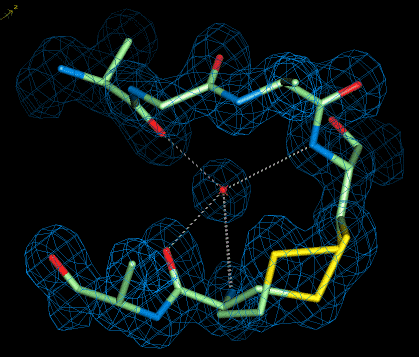
We solved not only several ultrahigh-resolution structures of the left-handed Z-DNA duplex, but also of a DNA/RNA hybrid (duplex formed from DNA and RNA chains) and DNA|RNA chimera (alternating deoxy- and ribo-nucleotides). In addition we determined the structure of several fiendishly twinned crystals and of a centrosymmetric DNA crystal, formed from a racemic mixture of DNA molecules with D- and L-deoxyriboses. Recently, we have studied a series of RNA duplexes with a tandem of GA mismatches in the middle, flanked by different combinations (polarity) of CG and iCiG base pairs.
Photosynthesis-related proteins


RbcX is a chaperone assisting in the assembly of RuBisCO, the CO2 fixing complex that is the most
abundant protein on Earth. The dimeric RbcX protein has a boomerang shape with alpha-helix bundles on both ends and a pair of very
long helices reaching from one bundle to the other. The long helices have a very strong kink in the middle, which - according to
normal-mode analysis - is a hinge for a large-amplitude butterfly motion of the molecule. It is very likely that this "breathing"
motion of the RbcX molecule is connected with binding and release of the RuBisCO large subunit (RbcL) during RuBisCO assembly. c6
is a special cytochrome found in algae, cyanobacteria and plants where it acts in electron transfer between cytochrome b6f and
photosystem I (PSI). c6 has a covalently bound heme residue and is characterized by a very high redox potential of nearly 0.4 V.
We have determined atomic-resolution structures of c6 from a mesophilic cyanobacterium (Synechococcus) in the reduced and
oxidized states.
Macromolecular structure at ultrahigh resolution


The crystal structure of a mutant of BPTI
(Bovine Pancreatic Trypsin Inhibitor) has been refined using low-temperature
synchrotron data extending to 0.86-A resolution to an R-factor of 0.1035.
From full-matrix least-squares refinement the accuracy of C-C bond distances
is 0.010 A. Many H-atoms could be seen in difference electron density maps,
including those in water molecules. The structure reveals a double-conformation
C14-C38 disulfide bridge and a salt bridge between the N- and C-termini.
It also provides examples of asparagine residues in unusual conformations
and illustrates the importance of N-H...pi hydrogen bonds in stabilizing
the molecular structure. As a result of the high resolution and high quality
of the electron-density maps, about 20% of all residues are found, and
successfully modeled, in alternate conformations. At this resolution the
refinement is highly overdetermined allowing for removal of main-chain
restraints and for unbiased verification of the stereochemical standards
used in protein structure refinements at lower resolution. This reveals,
for instance, that deviations up to 20 degrees from peptide bond planarity
are quite possible. Our crystal structure of Z-DNA refined at 0.53 A resolution
to R=0.067 without restraints, shows unusual regularity of the left-handed DNA
duplex in the absence of metal cations and indicates the need to revise some of the stereochemical standards
used in nucleic acid refinement at lower resolution. We have created a webserver RestraintLib that provides revised
restraints for nucleic acid refinement.
Crystallographic methodology


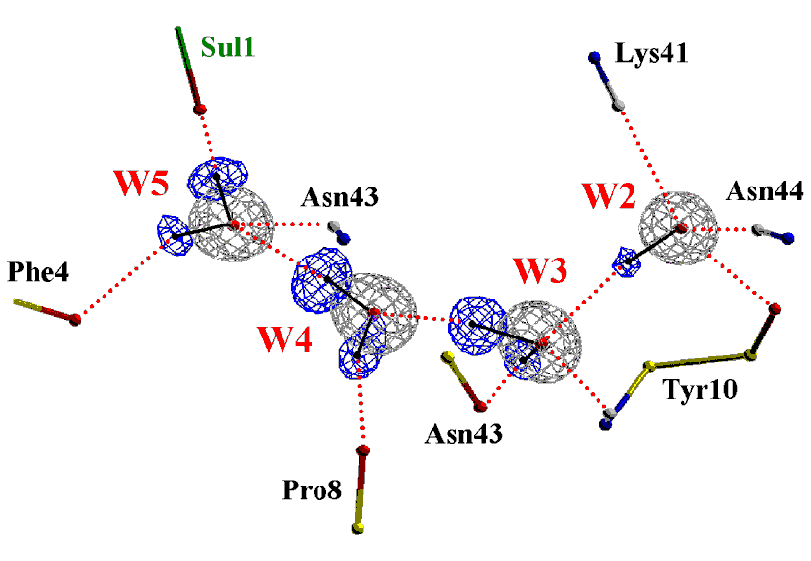
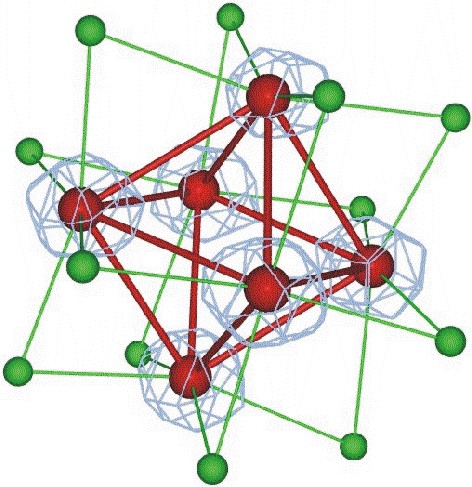
The crystal structure of a Z-DNA hexamer has been solved from the anomalous signal of the P atoms at copper wavelength. The multiplicity of the diffraction data was the most crucial single factor for the solution of the phase problem. The structure was refined to an R factor of 0.089 at 0.95 A resolution. In another study, we have demonstrated the power of the (Ta6Br12)2+ complex for phasing protein structures and determined the precise geometry and molecular interactions of this cation in a protein crystal context. The crystals of Hyp-1 in complex with ANS turned out to have commensurately modulated superstructure with 28 protein molecules in the asymmetric unit. The structure of this crystal (which also showed tetartohedral twinning) was solved (in collaboration with Prof. Randy Read, Univ. of Cambridge) by Molecular Replacement despite extremely complex, seven-fold, translational Non-Crystallographic Symmetry (tNCS). Modulated crystal structures are extremely rare in macromolecular crystallography. We are also developing Internet tools for teaching crystallography, available at this link (including a PXQuiz) and here. We also harnessed artificial intelligence to recognize ligands in the electron density of macromolecular structures. In view of the explosive growth of the PDB, we used this huge volume of examples to train a computer algorithm to carry out ligand recognition automatically. This machine learning exercise has indeed worked and the algorithm that has been developed, called CheckMyBlob, is now able to successfully identify examples that it has not seen before. Moreover, the algorithm will be able to continually improve, as the dataset of past examples continues to grow.
Crystal engineering
Our interest in this area is on hydrogen bonds as determinants of supramolecular organization, with focus on both extremely strong (i.e. short) and very weak hydrogen bonds. In the latter category, we are interested in C-H...X and Y-H...pi interactions. We have discovered interaction mimicry utilizing C-H...O bonds and leading to one-dimensional isostructurality. Also, we have used the N-H...N and C-H...O synthons to engineer a supramolecular helix. When applied to nucleoside salts, the concept of supramolecular synthons has revealed pre-ferred interaction patterns, also including C-H donors. Our current focus is on co-crystals as potential vehicles for Active Pharmaceutical Ingredients (APIs).
Supramolecular biocrystallography

We are using Peptidic Organic Frameworks (POFs) to engineer suitable supramolecular environment for protein crystallization. The cornerstone of our POFs is a resorcinarene macrocycle decorated with ologopeptidic tentacles. Such molecules spontaneously dimerize via beta-sheet interactions between their tentacles, creating a hollow capsule that can be loaded with a suitable cargo, e.g. a fullerene molecule. By decorating the resorcinarene scaffold with different functional groups on the other side, we hope to achieve spontaneous formation of 3D frameworks with controllable voids, designed for accommodation of a specific macromolecule. The capsules are interesting in their own right as molecular containers that can be used, for example, for drug encapsulation and delivery, or as interesting nanomaterials.
Last update: February 12, 2021 (MJ)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}